


КИСЛOТНО-ОСНОВНOЙ КАТАЛИЗ (кислотно-основный катализ), ускорение хим. р-ций в присут. к-т и оснований. В качестве катализаторов используют: в гомог. кислотном катализе протонные к-ты (H2SO4, HCl, Н3РО4, CH3C6H4SO2OH и др.) в воде и водно-орг. р-рителях, апротонные к-ты (АlС13, BF3, SnCl4 и др.) в неводных р-рителях, сверхкислоты (HF SbF5, HSO3F SbF5 и др.) в неводных р-рителях; в гетерог. кислотном катализе прир. глины, аморфные и кристаллич. алюмосиликаты, фосфорную и полифосфорные к-ты, нанесенные на носитель, катиониты; в гомог. основном катализе оксиды щелочных металлов, амины в воде, орг. и водно-орг. р-рителях; в гетерог основном катализе - оксиды металлов (CaO, MgO и др.).
При К.-о. к. в большинстве случаев из реагентов и катализатора в равновесных стадиях образуются реакционноспособные (а также нереакционноспособные) комплексы разл. состава, к-рые являются ионизир. формой реагента. В лимитирующих стадиях комплексы превращ. в продукты р-ции. В случае р-ции с одним реагентом (Р) эти стадии выражаются ур-нием:
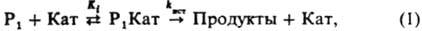
где Кат-разл. формы катализатора: недиссоциир. к-ты или основания, ионы или ионные пары, образующиеся при их ионизации; P1 Кат-комплексы; Кi - константа равновесия образования комплексов Р1 Кат; kист - элементарная константа скорости превращ. комплексов в продукты. В случае р-ции двух реагентов (P1+Р2: Продукты; Р2+Р2: Продукты) реакционноспособный комплекс образуется, как правило, из комплекса одного реагента с несвязанным в комплекс др. реагентом:
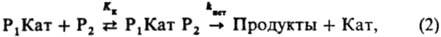
где Кк - константа равновесия образования комплексов Р1 Kат Р2. Константы равновесия Ki и Кк выражают ур-ниями:
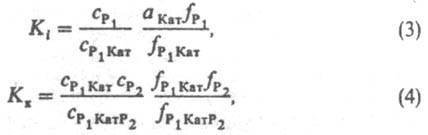
где (здесь и далее) а, с, f-соотв. термодинамич. активность, концентрация и коэф. активности реагирующих частиц; aKатfP1/fP1Кат - ф-ция, характеризующая способность среды переводить реагенты в комплексы РКат. При варьировании в широких пределах сКат реализуются (особенно в р-рах) неск. маршрутов р-ции - хим. превращений, приводящих к образованию одних и тех же продуктов через реакционноспособные комплексы (РК) разл. состава. В р-ре с постоянной сКат скорость р-ции по каждому маршруту Wi выражается ур-нием:
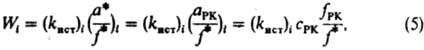
где (kист)i - элементарная константа скорости по i-му маршруту, а* и f* - относятся к активир. комплексу. Из опытных данных следует, что множитель fРК/f* не изменяется при варьировании сКат и его приравнивают к единице. В р-ре с постоянной сКат наблюдаемая скорость р-ции
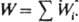
Экспериментально определяют эффективную константу скорости kэфф, характеризующую скорость р-ции при постоянной т-ре и наличии катализатора определенного состава. В ур-ниях для связи fэфф со св-вами катализатора учитывают все его комплексы с реагентом: реакционноспособные (РКат)РК и нереакционноспособные (РКат)НРК. Напр., при кислотном гидролизе амидов, анилидов ионизированная по атому азота форма является реакционноспособной, ионизированная по карбонильной группе - либо намного менее реакционноспособна, либо нереакционноспособна. Для описания влияния ионизирующих св-в среды на kэфф совместно решают ур-ние материального баланса для текущих концентраций реагентов и ур-ния (3) и (5) в случае р-ции одного реагента и дополнительно ур-ние (4) для р-ции двух реагентов. напр. для р-ции, протекающей по схеме (2):

В случае р-ции двух реагентов, если один (Р2) является компонентом р-рителя (напр., Н2О при гидролизе в водных р-рах), при условии ср2>>ер1 вычисляют эффективную константу скорости первого порядка, включающую термодинамич. активность р-рителя. В р-циях Рх с Р2 в р-рах к-т и оснований реакционноспособный комплекс состава P1KaтP2 может получаться и в лимитирующей стадии из ионизир. формы одного реагента и неионизир. формы другого:
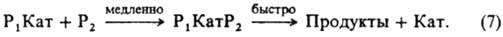
В этом случае для описания влияния ионизирующих св-в среды на kэфф пригодно ур-ние (6), при замене величины (fP2сP2)/(КкfP1КатP2) на термодинамич. активность растворителя. Поскольку крайне редко удается установить прямыми физ. методами состав и концентрацию промежут. комплексов, в настоящее время основным методом установления их состава, детального механизма р-ции и вычисления величин Кi и Кк является кинетич. метод, основанный на применении ур-ний типа (6) для объяснения наблюдаемой зависимости kэфф от ионизирующей способности среды. Для этого измеряют kэфф в широком диапазоне сКат в р-ре и подбирают ф-ции аКат fP/fPКат для описания влияния среды на kэфф. наиб. исследованы каталитич. св-ва сильных к-т в водных р-рах. Ионизирующая способность таких р-ров обусловлена образованием комплексов реагента: 1) с сольватированным протоном и 2) с недиссоциированной к-той. К первым относятся протонир. форма реагента и ее комплексы с р-рителем. В этом случае аКатfP/fPKат выражают (для водных р-ров) ф-циями h0 =aH3O+fP/fPH+, h0aH2O и h0a2Н2O, где h0 - измеренная индикаторным методом кислотность среды, fРН+ -коэф. активности протонир. формы реагента. Ко вторым относятся комплексы реагента с к-той (НА) и ее гидратами. При этом aКатfР/fРКат кат принимает значения аНА, aНАaН2O и др. При гидролизе нек-рых карбонильных соед. aКатfP/fРКат=cH5O2+ Однозначную информацию о составе реакционноспособных комплексов, как правило, получают на основании данных по изменению kэфф в р-рителе постоянного состава при варьировании сКат в широких пределах в условиях малой степени связывания реагента в комплексы. наиб. простые зависимости получают для р-ции одного реагента, когда он равновесно связывается только в один реакционноспособный комплекс. В таком случае ур-ние (4) упрощается:
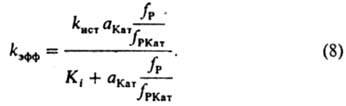
Опытным путем установлено, что в водных р-рах сильных к-т kист не зависит от сКат. Кi, по определению, не должно зависеть от сКат. Вопрос о влиянии состава смешанного р-рителя на kист пока достаточно не исследован. В условиях, когда Ki и (aКатfР)/fРКат соизмеримы, по ур-нию (8) вычисляют раздельно kист и Ki. При аКатfР/fРКат >> Ki величина kэфф=kист; при kКатfP/fРКат<<Ki имеет место соотношение:
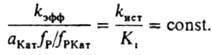
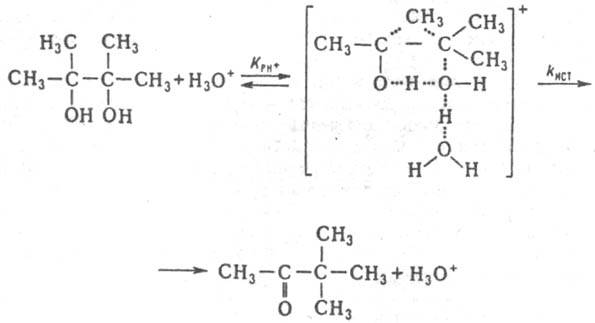
Ниже приведен пример, раскрывающий природу каталитич. действия водных р-ров к-т в условиях, когда реакционноспособный комплекс образуется и превращ. в продукты согласно схеме (1) при условии, что аКатfP/fРКат<<Ki. Ионизирующая способность среды из-за превращ. реагента в протонир. форму и ее комплексы с Н2О проявляется при наличии зависимостей Кэфф/h0=const, kэфф(h0aH2О)=const kэфф/(h0a2H2O)=const. Если в р-ции kэфф/h0=const (lgkэфф+H0=const, где H0=-lgh0 - ф-ция кислотности Гаммета), то aкатfP/fКат=h0 и реакционноспособным комплексом является протонир. форма реагента РН3О+. Такая закономерность имеет место при проведении пинаколиновой перегруппировки в конц. H2SO4:
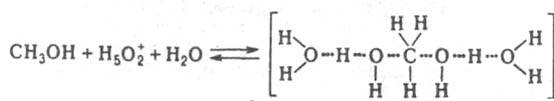
При варьировании сKaт значение kэфф/(h0аH2O) постоянно в р-циях дегидратации, напр. 2-фенил-2-пропанола в серной к-те, 2-метил-2-бутанола в азотной к-те. Следовательно, аKатfP/fPKaт равно h0аH2O и реакционноспособным является комплекс молекулы воды с протонир. формой реагента. Имеется и пример постоянства kэфф/(h0a2H2O), что наблюдается в р-ции изотопного обмена атома кислорода в СН3ОН, протекающего в серной и хлорной к-тах. В этом случае аKaт/fP/fРКaт=h0a2H2O и реакционноспособным является симметричный комплекс:
Ф-ции h0, h0аH2O и h0а2H2O стандартизованы к воде, и в сильно разбавленных р-рах сильных к-т они численно равны концентрации ионов Н5О+2 в моль/л. В таких р-рах для приведенных выше р-ций при варьировании сKaт должно соблюдаться постоянство значений kэфф/cH2O+. При гидролизе нек-рых амидов, анилидов, сложных эфиров kэфф/cH2O+ постоянно не только в разбавленных, но и в конц. водный р-рах НСlO4, H2SO4. Если гидролиз протекает по схеме (1), то реакционноспособный комплекс образуется из реагента и иона Н5О2+, а выраженная в концентрациях константа равновесия KH5O2+; не изменяется при варьировании сКат:
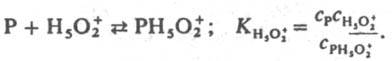
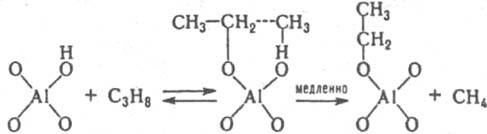
Не исключено, что kэфф/cH2O+=const из-за того, что р-ция протекает по схеме (7) Ионизирующая способность среды из-за комплексообразования реагента с молекулами недиссоциир. сильной к-ты и ее гидратом проявляется при наличии зависимостей kэфф/aHA=const, kэфф/(aHAaH2O)=const и др. В первом случае aKaт=const и реакци онноспособный комплекс имеет состав РИА; такая закономерность, напр., реализуется при дегидратации b-фенил-b-гидроксипропионовой к-ты и 2-(4-мето-ксифенил)-2-пропанола в серной к-те. Во втором случае акатfP/fPKат=аHАаH2O и комплекс имеет состав РНАН2О. Гидрат НА.Н2О можно рассматривать и как ионную пару Н3О+А-. Каталитич. активность гидратов сильных к-т проявляется в р-циях дегидратации 1-фенилэтанола в хлорной к-те, гидратации цис-коричной к-ты и рацемизации b-фенил-b-гидроксипропионовой к-ты в серной к-те. Согласно ур-нию (6), должен наблюдаться максимум kэфф в зависимости от сКат, если наряду с реакционноспособными образуются и нереакционноспособные комплексы и при увеличении сКат значение aКат/fP/fPKат для нeрeакционноспособных комплексов возрастает быстрее и более резко, чем для реакционноспособных. Напр., если aКатfP/fPKaт равно сH5O2+ для реакционноспособных комплексов и равно h0, либо h0aH2O для нереакционноспособных. В водных р-рах слабых к-т каталитич. активность проявляют ионы и недиссоциир. молекулы к-ты. Взаимосвязь между каталитич. активностью слабых к-т и их константами диссоциации в нек-рых случаях удается описать корреляц. ур-нием Брёнстеда. В сильно разбавленных водных р-рах оснований ндблюдается постоянство kэфф/cOH - при варьировании сOH-. Обоснованных количеств. характеристик ионизирующей способности умеренно конц. и конц. р-ров оснований пока нет. Добавление солей к водным р-рам к-т и оснований существенно изменяет их каталитич. активность; в случае слабых к-т и оснований изменяется степень их диссоциации на ионы из-за наличия общего иона и увеличения ионной силы р-ра. В случае сильных к-т изменяются величины h0, аНА, аН2O, что приводит к увеличению относит, концентрации комплексов, образующихся из частиц Н3О+ А-, НА, а также усилению реакц. способности ионов Н5О2+, напр. при гидролизе в условиях, когда aКатfP/fPKат=сH2O+. В случае сильных оснований катионы металлов усиливают каталитич. действие сольватир. ионов ОН-. Лимитирующие стадии катализируемых к-тами и основаниями р-ций часто осуществляются по синхронным механизмам. В неводных р-рителях к-ты и основания образуют преим. мол. комплексы. Каталитич. действие апротонных к-т обусловлено образованием из них и реагентов мол. реакционно-способных комплексов. Катиониты в Н-форме и нанесенные кислотные катализаторы обладают каталитич. активностью гл. обр. из-за наличия жидкой фазы, содержащей к-ту. Нанесенные кислотные катализаторы готовят нанесением на носитель (силикофосфат SiO2.Р2О5, силикагель, кварц, фосфаты) разл. жидких к-т, гл. обр. фосфорной. Каталитич. активность таких катализаторов зависит от кол-ва нанесенной к-ты на 1 г носителя и ее концентрации в жидкой фазе, зависящей от равновесного давления водяного пара над катализатором. Для характеристики ионизирующих св-в нанесенных кислотных катализаторов можно использовать такие же ф-ции аКaтfP/fPKaт, как и для р-ров. Специфич. св-во практически всех гетерог. кислотно-основных катализаторов наличие на их пов-сти центров разл. силы (кислотной или основной), что обусловлено неоднородностью пов-сти твердых тел. Из-за наличия подобных центров образуются сильно поляризованные комплексы реагента с катализатором. Энергетически выгодными являются синхронно протекающие лимитирующие стадии. Так, крекинг парафинов на Аl2О3 может протекать по схеме:
Лит. Гам мет Л , Основы физической органической химии, пер. с англ., М., 1972; Танабе К . Твердые кислоты и основания, пер. с англ., М., 1973, Белл Р , Протон в химии, пер. с англ., М., 1977; Виниик М И , "Кинетика и катализ", 1980. т 21. в. I.e. 136 58. Казанский В Б . там же. 1987. т 28. в. I, с. 47 60 МИ Винник
где Кат-разл. формы катализатора: недиссоциир. к-ты или основания, ионы или ионные пары, образующиеся при их ионизации; P1 Кат-комплексы; Кi - константа равновесия образования комплексов Р1 Кат; kист - элементарная константа скорости превращ. комплексов в продукты. В случае р-ции двух реагентов (P1+Р2: Продукты; Р2+Р2: Продукты) реакционноспособный комплекс образуется, как правило, из комплекса одного реагента с несвязанным в комплекс др. реагентом:
где Кк - константа равновесия образования комплексов Р1 Kат Р2. Константы равновесия Ki и Кк выражают ур-ниями:
где (здесь и далее) а, с, f-соотв. термодинамич. активность, концентрация и коэф. активности реагирующих частиц; aKатfP1/fP1Кат - ф-ция, характеризующая способность среды переводить реагенты в комплексы РКат. При варьировании в широких пределах сКат реализуются (особенно в р-рах) неск. маршрутов р-ции - хим. превращений, приводящих к образованию одних и тех же продуктов через реакционноспособные комплексы (РК) разл. состава. В р-ре с постоянной сКат скорость р-ции по каждому маршруту Wi выражается ур-нием:
где (kист)i - элементарная константа скорости по i-му маршруту, а* и f* - относятся к активир. комплексу. Из опытных данных следует, что множитель fРК/f* не изменяется при варьировании сКат и его приравнивают к единице. В р-ре с постоянной сКат наблюдаемая скорость р-ции
Экспериментально определяют эффективную константу скорости kэфф, характеризующую скорость р-ции при постоянной т-ре и наличии катализатора определенного состава. В ур-ниях для связи fэфф со св-вами катализатора учитывают все его комплексы с реагентом: реакционноспособные (РКат)РК и нереакционноспособные (РКат)НРК. Напр., при кислотном гидролизе амидов, анилидов ионизированная по атому азота форма является реакционноспособной, ионизированная по карбонильной группе - либо намного менее реакционноспособна, либо нереакционноспособна. Для описания влияния ионизирующих св-в среды на kэфф совместно решают ур-ние материального баланса для текущих концентраций реагентов и ур-ния (3) и (5) в случае р-ции одного реагента и дополнительно ур-ние (4) для р-ции двух реагентов. напр. для р-ции, протекающей по схеме (2):
В случае р-ции двух реагентов, если один (Р2) является компонентом р-рителя (напр., Н2О при гидролизе в водных р-рах), при условии ср2>>ер1 вычисляют эффективную константу скорости первого порядка, включающую термодинамич. активность р-рителя. В р-циях Рх с Р2 в р-рах к-т и оснований реакционноспособный комплекс состава P1KaтP2 может получаться и в лимитирующей стадии из ионизир. формы одного реагента и неионизир. формы другого:
В этом случае для описания влияния ионизирующих св-в среды на kэфф пригодно ур-ние (6), при замене величины (fP2сP2)/(КкfP1КатP2) на термодинамич. активность растворителя. Поскольку крайне редко удается установить прямыми физ. методами состав и концентрацию промежут. комплексов, в настоящее время основным методом установления их состава, детального механизма р-ции и вычисления величин Кi и Кк является кинетич. метод, основанный на применении ур-ний типа (6) для объяснения наблюдаемой зависимости kэфф от ионизирующей способности среды. Для этого измеряют kэфф в широком диапазоне сКат в р-ре и подбирают ф-ции аКат fP/fPКат для описания влияния среды на kэфф. наиб. исследованы каталитич. св-ва сильных к-т в водных р-рах. Ионизирующая способность таких р-ров обусловлена образованием комплексов реагента: 1) с сольватированным протоном и 2) с недиссоциированной к-той. К первым относятся протонир. форма реагента и ее комплексы с р-рителем. В этом случае аКатfP/fPKат выражают (для водных р-ров) ф-циями h0 =aH3O+fP/fPH+, h0aH2O и h0a2Н2O, где h0 - измеренная индикаторным методом кислотность среды, fРН+ -коэф. активности протонир. формы реагента. Ко вторым относятся комплексы реагента с к-той (НА) и ее гидратами. При этом aКатfР/fРКат кат принимает значения аНА, aНАaН2O и др. При гидролизе нек-рых карбонильных соед. aКатfP/fРКат=cH5O2+ Однозначную информацию о составе реакционноспособных комплексов, как правило, получают на основании данных по изменению kэфф в р-рителе постоянного состава при варьировании сКат в широких пределах в условиях малой степени связывания реагента в комплексы. наиб. простые зависимости получают для р-ции одного реагента, когда он равновесно связывается только в один реакционноспособный комплекс. В таком случае ур-ние (4) упрощается:
Опытным путем установлено, что в водных р-рах сильных к-т kист не зависит от сКат. Кi, по определению, не должно зависеть от сКат. Вопрос о влиянии состава смешанного р-рителя на kист пока достаточно не исследован. В условиях, когда Ki и (aКатfР)/fРКат соизмеримы, по ур-нию (8) вычисляют раздельно kист и Ki. При аКатfР/fРКат >> Ki величина kэфф=kист; при kКатfP/fРКат<<Ki имеет место соотношение:
Ниже приведен пример, раскрывающий природу каталитич. действия водных р-ров к-т в условиях, когда реакционноспособный комплекс образуется и превращ. в продукты согласно схеме (1) при условии, что аКатfP/fРКат<<Ki. Ионизирующая способность среды из-за превращ. реагента в протонир. форму и ее комплексы с Н2О проявляется при наличии зависимостей Кэфф/h0=const, kэфф(h0aH2О)=const kэфф/(h0a2H2O)=const. Если в р-ции kэфф/h0=const (lgkэфф+H0=const, где H0=-lgh0 - ф-ция кислотности Гаммета), то aкатfP/fКат=h0 и реакционноспособным комплексом является протонир. форма реагента РН3О+. Такая закономерность имеет место при проведении пинаколиновой перегруппировки в конц. H2SO4:
При варьировании сKaт значение kэфф/(h0аH2O) постоянно в р-циях дегидратации, напр. 2-фенил-2-пропанола в серной к-те, 2-метил-2-бутанола в азотной к-те. Следовательно, аKатfP/fPKaт равно h0аH2O и реакционноспособным является комплекс молекулы воды с протонир. формой реагента. Имеется и пример постоянства kэфф/(h0a2H2O), что наблюдается в р-ции изотопного обмена атома кислорода в СН3ОН, протекающего в серной и хлорной к-тах. В этом случае аKaт/fP/fРКaт=h0a2H2O и реакционноспособным является симметричный комплекс:
Ф-ции h0, h0аH2O и h0а2H2O стандартизованы к воде, и в сильно разбавленных р-рах сильных к-т они численно равны концентрации ионов Н5О+2 в моль/л. В таких р-рах для приведенных выше р-ций при варьировании сKaт должно соблюдаться постоянство значений kэфф/cH2O+. При гидролизе нек-рых амидов, анилидов, сложных эфиров kэфф/cH2O+ постоянно не только в разбавленных, но и в конц. водный р-рах НСlO4, H2SO4. Если гидролиз протекает по схеме (1), то реакционноспособный комплекс образуется из реагента и иона Н5О2+, а выраженная в концентрациях константа равновесия KH5O2+; не изменяется при варьировании сКат:
Не исключено, что kэфф/cH2O+=const из-за того, что р-ция протекает по схеме (7) Ионизирующая способность среды из-за комплексообразования реагента с молекулами недиссоциир. сильной к-ты и ее гидратом проявляется при наличии зависимостей kэфф/aHA=const, kэфф/(aHAaH2O)=const и др. В первом случае aKaт=const и реакци онноспособный комплекс имеет состав РИА; такая закономерность, напр., реализуется при дегидратации b-фенил-b-гидроксипропионовой к-ты и 2-(4-мето-ксифенил)-2-пропанола в серной к-те. Во втором случае акатfP/fPKат=аHАаH2O и комплекс имеет состав РНАН2О. Гидрат НА.Н2О можно рассматривать и как ионную пару Н3О+А-. Каталитич. активность гидратов сильных к-т проявляется в р-циях дегидратации 1-фенилэтанола в хлорной к-те, гидратации цис-коричной к-ты и рацемизации b-фенил-b-гидроксипропионовой к-ты в серной к-те. Согласно ур-нию (6), должен наблюдаться максимум kэфф в зависимости от сКат, если наряду с реакционноспособными образуются и нереакционноспособные комплексы и при увеличении сКат значение aКат/fP/fPKат для нeрeакционноспособных комплексов возрастает быстрее и более резко, чем для реакционноспособных. Напр., если aКатfP/fPKaт равно сH5O2+ для реакционноспособных комплексов и равно h0, либо h0aH2O для нереакционноспособных. В водных р-рах слабых к-т каталитич. активность проявляют ионы и недиссоциир. молекулы к-ты. Взаимосвязь между каталитич. активностью слабых к-т и их константами диссоциации в нек-рых случаях удается описать корреляц. ур-нием Брёнстеда. В сильно разбавленных водных р-рах оснований ндблюдается постоянство kэфф/cOH - при варьировании сOH-. Обоснованных количеств. характеристик ионизирующей способности умеренно конц. и конц. р-ров оснований пока нет. Добавление солей к водным р-рам к-т и оснований существенно изменяет их каталитич. активность; в случае слабых к-т и оснований изменяется степень их диссоциации на ионы из-за наличия общего иона и увеличения ионной силы р-ра. В случае сильных к-т изменяются величины h0, аНА, аН2O, что приводит к увеличению относит, концентрации комплексов, образующихся из частиц Н3О+ А-, НА, а также усилению реакц. способности ионов Н5О2+, напр. при гидролизе в условиях, когда aКатfP/fPKат=сH2O+. В случае сильных оснований катионы металлов усиливают каталитич. действие сольватир. ионов ОН-. Лимитирующие стадии катализируемых к-тами и основаниями р-ций часто осуществляются по синхронным механизмам. В неводных р-рителях к-ты и основания образуют преим. мол. комплексы. Каталитич. действие апротонных к-т обусловлено образованием из них и реагентов мол. реакционно-способных комплексов. Катиониты в Н-форме и нанесенные кислотные катализаторы обладают каталитич. активностью гл. обр. из-за наличия жидкой фазы, содержащей к-ту. Нанесенные кислотные катализаторы готовят нанесением на носитель (силикофосфат SiO2.Р2О5, силикагель, кварц, фосфаты) разл. жидких к-т, гл. обр. фосфорной. Каталитич. активность таких катализаторов зависит от кол-ва нанесенной к-ты на 1 г носителя и ее концентрации в жидкой фазе, зависящей от равновесного давления водяного пара над катализатором. Для характеристики ионизирующих св-в нанесенных кислотных катализаторов можно использовать такие же ф-ции аКaтfP/fPKaт, как и для р-ров. Специфич. св-во практически всех гетерог. кислотно-основных катализаторов наличие на их пов-сти центров разл. силы (кислотной или основной), что обусловлено неоднородностью пов-сти твердых тел. Из-за наличия подобных центров образуются сильно поляризованные комплексы реагента с катализатором. Энергетически выгодными являются синхронно протекающие лимитирующие стадии. Так, крекинг парафинов на Аl2О3 может протекать по схеме:
Лит. Гам мет Л , Основы физической органической химии, пер. с англ., М., 1972; Танабе К . Твердые кислоты и основания, пер. с англ., М., 1973, Белл Р , Протон в химии, пер. с англ., М., 1977; Виниик М И , "Кинетика и катализ", 1980. т 21. в. I.e. 136 58. Казанский В Б . там же. 1987. т 28. в. I, с. 47 60 МИ Винник