


НИТРОВАНИЕ, введение
нитрогруппы —NO2 в молекулы орг. соединений. Может проходить по электроф.,
нуклеоф. и радикальному механизмам; активные частицы в этих р-циях-соотв. катион
нитрония NO2, нитрит-ион NO2 и радикал NO2.
H. может осуществляться по атомам С, N, О замещением атома водорода (прямое
Н.) или др. функц. групп (заместительное Н.) либо в результате присоединения
группы NO2 по кратной связи.
Электрофильное Н. Среди
электроф. нитрующих агентов доминирующее положение занимает HNO3.
Безводная и конц. HNO3 способны к самопротонированию: 2HNO3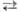 [Н2NО3]+ + NO3-
[Н2NО3]+ + NO3- 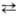 NО2+
+ NO-3 + H2O. Присутствие воды снижает концентрацию
NO+2 и в 93- 95%-ной HNO3 спектрофотометрически
он уже не обнаруживается. Для увеличения нитрующей активности HNO3
используют ее смеси с H2SO4 или олеумом, к-рые генерируют
NO2, связывая воду:
NО2+
+ NO-3 + H2O. Присутствие воды снижает концентрацию
NO+2 и в 93- 95%-ной HNO3 спектрофотометрически
он уже не обнаруживается. Для увеличения нитрующей активности HNO3
используют ее смеси с H2SO4 или олеумом, к-рые генерируют
NO2, связывая воду:
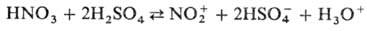
В безводной H2SO4
при содержании HNO3 меньше 10% равновесие полностью сдвинуто вправо.
Применяют также комбинации HNO3, разл. оксидов азота и орг. нитратов
с к-тами Льюиса (АlСl3, ZnCl2, BF3 и др.);
сильным нитрующим действием обладает смесь HNO3 с (СН3СО)2О
благодаря образованию ацетилнитрата и N2O5 (последний
при содержании в смеси более 90% HNO3 полностью диссоциирует на NO+2
и NO-3); перспективны также смеси HNO3 с безводным
SO3 или N2O5. Вместо HNO3 можно
применять ее соли, однако в пром-сти такой метод не используют из-за осложнения
процесса регенерации отработанных к-т. В случае слабой взаимной р-римости нитрующего
агента и субстрата, а также для уменьшения побочных процессов Н. проводят в
орг. р-рителях, напр. нитрометане, сульфолане, уксусной к-те; полярные р-рители
способствуют диссоциации [H2NO3]+ и тем самым
увеличивают концентрацию NO2.
В лаб. практике широко
используют апротонные нитрующие агенты (нитраты, соли нитрония, полинитросоед.
и др.), активность к-рых в р-циях электрофильного Н. увеличивается в ряду: AlkONO2
< (CH3)2C(CN)ONO2 < < RC(N02)3
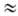 RN(N02)2
< NO2F < CH3COONO2 < < N2O5
< NO2+X-.
RN(N02)2
< NO2F < CH3COONO2 < < N2O5
< NO2+X-.
Субстратами для электрофильного
Н. служат ароматич. и гетероциклич. соед., олефины, относительно сильные СН-кислоты,
амины, спирты.
Н. ароматич. соед. протекает
по схеме:
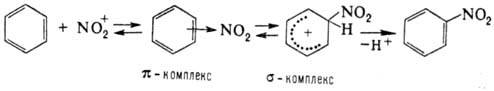
Возможно также образование
s-комплекса, в к-ром группа NO2 связана с атомом углерода кольца,
несущим заместитель (ипсо-атака). Соед. с электронодопорными заместителями более
реакционноспособны и нитруются в орто- и пара-положения, а с электроноакцепторными
- в мета-поло-жение. В пром-сти для Н. ароматич. соед. применяют в осн.
смесь HNO3 и H2SO4 (выход нитропродуктов ~
90-95%). Основная побочная р-ция - окисление, приводящее, как правило, к деструкции
ароматич. кольца. В зависимости от реакц. способности субстрата условия Н. варьируют
в широких пределах-от
водной HNO3 при 0°С (обязательно присутствие оксидов азота) до
дымящей HNO3 в олеуме при повыш. т-рах. При низких т-рах с высокой
скоростью протекает Н. ароматич. соед. солями нитрония; при этом часто лимитирующая
стадия-скорость растворения соли нитрония. Используют также заместительное Н.-замещение
сульфо-, диазо- и др. функц. групп. Этим приемом пользуются, в частности, в
случаях, когда невозможно прямое Н. Н. олефинов апротонными нитрующими агентами
в зависимости от условий и строения реагентов может идти по разным направлениям,
включая отщепление Н+, присоединение элементов р-рителя и противоиона,
полимеризацию и др., напр.:
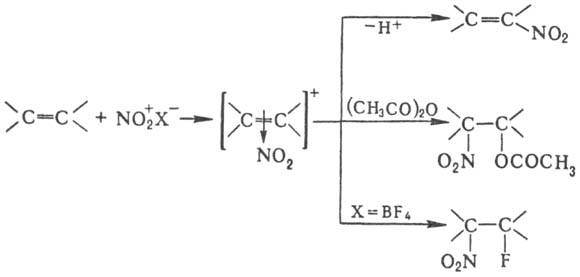
При Н. олефинов тетранитрометаном
в зависимости от строения олефина образуются либо алифатич. полинитро-соед.,
либо производные изоксазолидина, напр.:
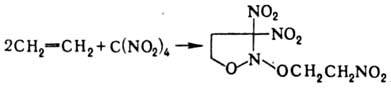
Нек-рые СН-кислоты при
Н. образуют анионы соответствующих нитросоед.; напр., при действии на флуорен
этил-нитрата в присут. С,Н5ОК образуется К-соль 9-нитро-флуорена,
примером Н. карбанионов может служить также превращ. солей моно- и динитросоед.
соотв. в геминальные ди- и тринитропроизводные при действии FNO2.
Соед. с активир. метиленовой
группой можно нитровать и в кислых условиях; напр., при обработке диэтилмалоната
HNO3 образуется нитродиэтилмалонат, Н. в аналогичных условиях 1,3-индандиона
с послед. щелочным гидролизом образующегося a-нитрокетона - удобный метод синтеза
первичных нитроалканов:
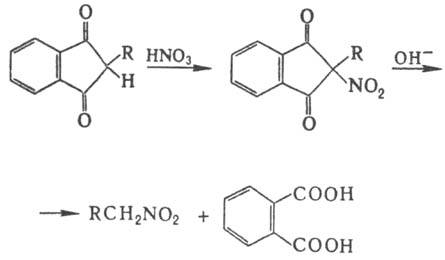
Электрофильное Н. аминов
в отличие от Н. по атому С-обратимый процесс и протекает по схеме:
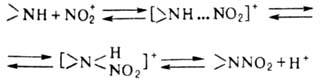
Лимитирующая стадия р-ции-перегруппировка
комплекса амина с NO2 в протонир. нитрамин.
В пром-сти Н. аминов проводят
кислыми нитрующими агентами (конц. HNO3 или ее смесями с H2SO4,
уксусной к-той или ангидридом). Слабоосновные амины и амиды нитруются с высокими
выходами. Высокоосновные амины (первичные и вторичные), протонир. форма к-рых
не реагирует с NO2+, превращают либо в амиды, к-рые нитруют
и затем снимают защитную ацильную группу щелочным гидролизом, либо в N-хлорамины;
в последнем случае Н. проводят в присут. катализаторов (НСl, ZnCl2).
Н. третичных аминов конц.
HNO3 или ее смесью с уксусным ангидридом сопровождается разрывом
связи С— N (такой тип Н. наз. нитролизом). Эту р-цию широко используют в пром-сти
ВВ, напр. для получения гексагена и октогена из уротропина. Жирно-ароматич.
амины типа ArNHR часто нитруются в ядро, что происходит в результате непосредственного
Н. по атому С или перегруппировки N-нитропроизводного; при этом группа NO2
вступает в ортo-положение к аминной функции. В ряде случаев Н. по атому
N проводят через стадию образования соли. Для этого амин обрабатывают разб.
HNO3 и на образовавшийся нитрат действуют конц. HNO3 или
уксусным ангидридом:
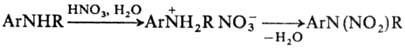
В лаб. условиях заместительное
Н. ацетамидов, сульфамидов, уретанов, имидов или их солей проводят в апротон-ной
среде апротонными нитрующими агентами, напр. солями нитрония:
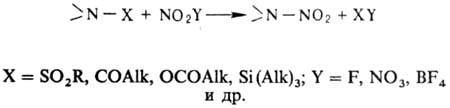
Из первичных аминов можно
синтезировать N,N-дини-троамины, к-рые, в свою очередь, являются нитрующими
агентами.
Спирты нитруют любыми нитрующими
агентами, содержащими NO+2 (в кислых средах р-ция обратима),
напр.: RCH2OH + NO2+X- 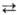 RCH2ONO2
+ НХ.
RCH2ONO2
+ НХ.
Нуклеофильное Н. осуществляют
солями HNO2:
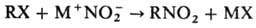
В р-цию вступают алкилгалогениды,
в осн. бромиды и иодиды (см. Мейера реакция), a-галогенкарбоновые к-ты
и их эфиры, алкилсульфаты. В качестве нитрующих агентов используют нитриты щелочных
металлов в апротонных диполярных р-рителях или проводят Н. в присут. краун-эфиров.
Побочные продукты р-ции-орг. нитриты, что связано с двойственной реакц. способностью
NO-2. Р-цию используют для получения алифатич. нитросоединений.
Радикальное Н. характерно
в осн. для парафинов и олефинов. Источником NO.2
служат HNO.3 и оксиды азота. Н. парафинов проводят
разб. HNO3 под давлением при повыш. т-ре (Коновалова реакция).
Р-ция Н. протекает по схеме:
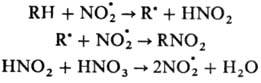
Наряду с Н. идет также
процесс окисления, связанный с взаимод. NO.2 с
орг. радикалом по атому кислорода. Наиб. легко протекает Н. по третичным атомам
углерода, трудно-по первичным. В пром-сти нитропарафины получают жидкофазным
и парофазным Н. смеси парафинов. Жидко-фазное Н. проводят HNO3 при
норм. или повыш. давлении и т-ре выше 180°С, или оксидами азота при давлении
2-4,5 МПа, 150-220 °С, время контакта ~15 с. В этих условиях линейные углеводороды
нитруются быстрее, чем их разветвленные
изомеры. Парофазное Н. (метод Хэсса) осуществляют HNO3 при давлении
0,7-1,0 МПа, 400-500 °С, время контакта ~ 1 с. Побочные процессы-деструкция
углеводородной цепи и окисление. Эти методы используют также для Н. алифатич.
боковых цепей жирно-ароматич. соед. (р-цию проводят в присут. катализаторов
-О2, О3, галогенов и др.),
Н. непредельных соед. HNO3
приводит к формальному замещению атома водорода у sp2-гибридизованного
атома углерода на группу NO2. Условия Н. зависят от строения непредельных
соединений. Обычно применяют 70-80%-ную HNO3 или разб. HNO3
в присут. оксидов азота.
Для Н. алкенов, циклоалкенов,
диалкил- и диарилацети-ленов можно использовать N2O4,
последний присоединяется по двойной связи, образуя вицинальные динитросоед.,
b-нитронитриты и b-нитронитраты, к-рые обычно легко отщепляют HNO2
или HNO3, давая непредельное нитро-соединение. Р-ция с ацетиленами
приводит к смеси вици-нальных цис- и транc-динитросоед. наряду
с продуктами окисления и деструкции.
По анион-радикальному механизму
проходит Н. тетра-нитрометаном солей мононитросоед. в гем-динитроалканы,
а также синтез последних из a-галогеннитроалканов при действии нитритов в щелочной
среде (р-ция Тер Меера):
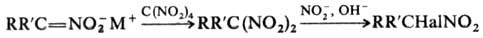
Р-ция Н. известна в орг.
химии с 1834 (синтез нитробензола Н. бензола азотной к-той, Э. Мичерлих). С
сер. 19 в. она используется в пром-сти в связи с открытием Н. Н. Зининым восстановления
нитробензола в анилин (см. Зинина реакция). Н.-наиб. удобный метод образования
связей С—N и N—N в молекулах орг. соед., широко используется в орг. синтезе.
По р-ции Н. в мире производится ~ 1 млн. т разл. нитропродуктов (гл. обр. в
ряду ароматич. соед.).
Лит. см. при ст. Нитросоединения. В. А. Тартакоеский.