


РАСТВОРЫ НЕЭЛЕКТРОЛИТОВ,
бинарные или многокомпонентные мол. системы, состав к-рых может изменяться непрерывным
образом (по крайней мере, в нек-рых пределах). В отличие от растворов электролитов,
в Р. н. (мол. р-рах) заряженные частицы в сколько-нибудь заметных концентрациях
отсутствуют. Р. н. могут быть твердыми, жидкими и газообразными. В данной статье
рассматриваются жидкие р-ры; см. также Твердые растворы.
Взаимная р-римость двух
жидкостей при заданных т-ре Т и давлении р м. б. полной (неограниченной)
или ограниченной. В последнем случае р-ры в нек-рой области составов расслаиваются,
т. е. разделяются на две жидкие фазы, отличающиеся по концентрации. В многокомпонентных
расслаивающихся р-рах число сосуществующих жидких фаз м. б. более двух. Если
один (или более) из компонентов Р.н. в чистом состоянии при заданных Т и
р является газом или твердым телом, область существования Р.н. простирается
от чистой жидкости (смеси жидкостей), выступающей в роли р-рителя, до состава,
отвечающего насыщ. р-ру.
Р.н. служат средой, в к-рой
протекают многие прир. и пром. процессы. Изучение и прогнозирование св-в этих
систем тесно связаны с такими практич. проблемами, как подбор р-рителей для
реализации технол. процессов, получение систем с заданными св-вами, разделение
прир. и пром. смесей (включая газы и нефти), глубокая очистка в-в.
Физ. химия изучает широкий
диапазон св-в р-ров. Наиб. разработана и имеет практически важные применения
равновесная термодинамика р-ров; дальнейший материал посвящен в осн. этому разделу
физ. химии р-ров. Кроме того, изучаются транспортные св-ва р-ров-диффузия, теплопроводность,
вязкость (см. Физико-химическая гидродинамика), а также спектроскопич.,
электрич., акустич. и др. физ. св-ва. Методы исследования макроскопич. св-в
Р. н. и их структурных характеристик во многом аналогичны методам исследования
индивидуальных жидкостей, но осн. внимание уделяется рассмотрению концентрац.
зависимостей св-в. Важнейшая задача физ.-хим. исследований - установление связи
между наблюдаемыми на опыте св-вами, структурой р-ров и характеристиками межмолекулярных
взаимодействий. Эксперим. информацию о структуре р-ров и межмолекулярных
взаимод. в них дают методы оптической и радиоспектроскопии, дифракционные, электрич.
и др. Важную роль в изучении Р.н. играет физико-химический анализ, основанный
на построении и исследовании фазовых диаграмм, концентрац. зависимостей термодинамич.
и др. физ. св-в (показателя преломления, вязкости, теплопроводности, акустич.
характеристик и др.). При этом одна из главных задач состоит в том, чтобы на
основании анализа диаграмм состав - свойство устанавливать факт образования
хим. соединений между компонентами Р.н. и находить их характеристики.
Значит. влияние на физ.
св-ва р-ров (в частности, на рассеяние света) оказывают флуктуации плотности,
концентрации, ориентации молекул. Роль флуктуации концентрации особенно велика
вблизи критич. точки р-римости (см. Критические явления).
Концентрационные зависимости
термодинамических функций. Особенностью термодинамич. описания Р. н. по сравнению
с чистыми компонентами является наличие дополнит. термодинамич. степеней свободы
системы, связанных с возможностью изменения состава системы (см. Фаз правило).
Число степеней свободы гомогенного n-компонентного р-ра равно n+1.
В качестве переменных, определяющих его состояние, наиб. удобно выбрать давление
р, т-ру Т и концентрации п — 1 компонентов. Состав Р. н.
чаще всего выражают через молярные доли компонентов xi, считая
независимыми переменными молярные доли всех компонентов, кроме n-го x1,...,
xn-1. Для задания концентрации используют и др. шкалы (молярности
с, моляльности т).
При описании концентрац.
зависимостей термодинамич. ф-ций важную роль играют парциальные молярные
величины Mi для i-го компонента, определяемые соотношением:
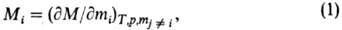
где М-любая экстенсивная
термодинамич. ф-ция (объем V, внутр. энергия U, энтальпия H,
энтропия S, энергии Гельм-гольца и Гиббса F и G, теплоемкость
Ср и т.д.), mi-число молей. Важнейшая парциальная
молярная величина -химический
потенциал mi (парциальная молярная энергия Гиббса); именно
через хим. потенциалы формулируются условия хим. и фазового равновесий в системе.
Концентрац. зависимость
термодинамич. св-в Р. н. нередко характеризуют функциями смешения Мт
- изменением термодинамич. ф-ции М при образовании р-ра из чистых
жидкостей. Рассматривают смешение при изотермо-изобар-ных (Т, р = const)
или изотермо-изохорных (Т, V = const) условиях, причем наиб. практич.
интерес представляет случай Т, р — const. Молярная ф-ция смешения при
этих условиях (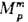 )
определена соотношением:
)
определена соотношением:
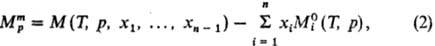
где 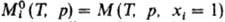 -молярное
значение ф-ции M
для чистой жидкости i при заданных Т и р. В частности,
молярная энергия Гиббса смешения
-молярное
значение ф-ции M
для чистой жидкости i при заданных Т и р. В частности,
молярная энергия Гиббса смешения
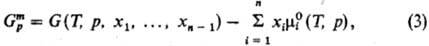
где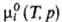 -хим.
потенциал чистой жидкости i при заданных Т
и р. Для чистых жидкостей
-хим.
потенциал чистой жидкости i при заданных Т
и р. Для чистых жидкостей  (рис., а). Поскольку образование
р-ра при смешении жидкостей - самопроизвольный процесс, то Gm
< 0.
(рис., а). Поскольку образование
р-ра при смешении жидкостей - самопроизвольный процесс, то Gm
< 0.
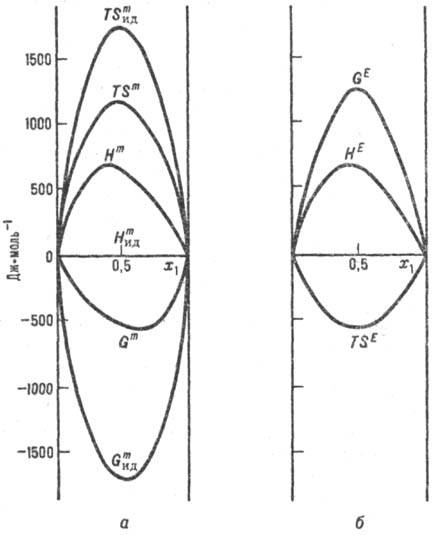
Функции смешения для реального
и идеального р-ров (а) и избыточные термо-динамич. функции (б). Система
пропанол - циклогексан, 298,5 К; x1-молярная доля пропанола.
Парциальная молярная ф-ция
смешения имеет вид:
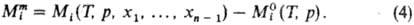
Термодинамич. соотношения,
связывающие величины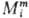 и
Мm между собой и с др. термодинамич. параметрами, аналогичны
соотношениям, связывающим Mi и М. Так,
и
Мm между собой и с др. термодинамич. параметрами, аналогичны
соотношениям, связывающим Mi и М. Так,
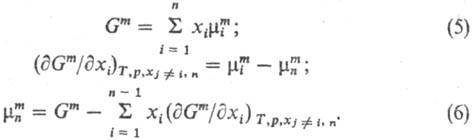
Зависимости энергии смешения
Гиббса и хим. потенциалов смешения от т-ры и давления выражаются соотношениями:
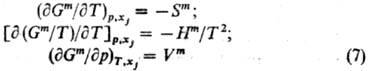
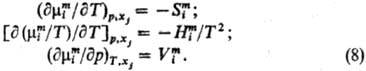
Вдали от критич.точки равновесия
жидкость-пар влияние давления на ф-ции смешения жидкого р-ра, как правило, незначительно
и им нередко пренебрегают.
Идеальные растворы. Имеется
неск. по существу идентичных определений идеального р-ра. Согласно одному из
наиб. удобных, это р-р, хим. потенциалы компонентов к-рого во всей областиконцентраций
отвечают ур-нию:
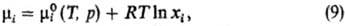
где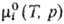 -хим.
потенциал чистой жидкости i при тех же давлении
и т-ре, что и рассматриваемый р-р, R-газовая постоянная. Ф-ции смешения
идеального р-ра:
-хим.
потенциал чистой жидкости i при тех же давлении
и т-ре, что и рассматриваемый р-р, R-газовая постоянная. Ф-ции смешения
идеального р-ра:
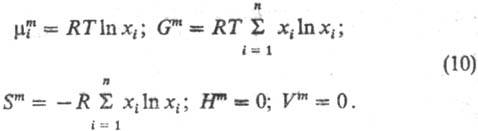
При образовании идеального
р-ра энергетич. изменений не происходит, изменяются только энтропия системы
и энтропийные составляющие термодинамич. ф-ций.
Если р-р идеальный, а равновесный
с ним пар ведет себя как идеальный газ, выполняется Рауля закон:

где pi-парциальное
давление i-го компонента над р-ром, 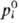 -давление насыщ. пара чистой жидкости при рассматриваемой т-ре.
-давление насыщ. пара чистой жидкости при рассматриваемой т-ре.
Идеальный р-р-гипотетич.
система, компоненты к-рой одинаковы по характеристикам межмол. взаимодействий.
Так, для бинарного р-ра одинаковыми должны быть потенциалы парного взаимод.
компонентов 1 и 2 u11, и22 и u12
(форма потенциалов м. б. любой; в частности, не исключается возможность
специфич. взаимод. компонентов 1-1, 2-2 и 1-2 одинаковой интенсивности).
При выполнении указанного требования смешение жидкостей не сопровождается энергетич.
изменениями. Т.к. число разл. конфигураций в
результате смешения увеличивается в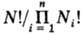 раз
(Ni- число
частиц i-го сорта, N = SNi), то энтропия
смешения
раз
(Ni- число
частиц i-го сорта, N = SNi), то энтропия
смешения 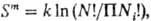 где
k-постоянная Больцмана; получаем ф-лы (10).
где
k-постоянная Больцмана; получаем ф-лы (10).
Поведение, близкое к идеальному,
обнаруживают р-ры, образованные молекулами разл. изотопного состава (напр.,
С6Н12 и C6D12), оптич. изомерами,
соседними гомологами с большой длиной цепи (напр., алканами С25 и
С26) и т.д.
Бесконечно разбавленные
Р. н. Р-р наз. бесконечно разбавленным по компоненту i, если xi:0.
Для определенности будем говорить о бинарном р-ре 1-2 при х2:0
(1-р-ри-тель, 2-растворенное в-во). Особенность такого р-ра состоит в
том, что молекулы растворенного в-ва окружены лишь молекулами р-рителя; взаимод.
между молекулами растворенного в-ва (типа 2-2)отсутствуют, существенны
лишь взаимод. типа 1-1 и 1-2. Пока р-р остается бесконечно разбавленным
и взаимод. 2-2 не проявляются, добавляемые молекулы 2 попадают в среднем
в одинаковые условия взаимод. с окружением и каждая дополнительно вносимая молекула
дает такой же вклад в среднюю энергию системы, объем и др. св-ва, как и предыдущие.
Парциальные молярные энергетич. характеристики р-ра остаются неизменными, и
концентрац. зависимость хим. потенциалов компонентов определяется чисто энтропийным
вкладом RTlnxi, как и в случае идеального р-ра. Т.
обр., при x2:0
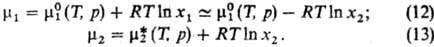
Здесь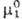 -хим.
потенциал чистого р-рителя. Стандартный хим. потенциал
-хим.
потенциал чистого р-рителя. Стандартный хим. потенциал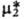 относится
к гицотетич. чистой жидкости 2, в к-рой компонент 2 обладает теми же парциальными
молярными энергетич.
св-вами, что и в бесконечно разб. р-ре.
относится
к гицотетич. чистой жидкости 2, в к-рой компонент 2 обладает теми же парциальными
молярными энергетич.
св-вами, что и в бесконечно разб. р-ре.
При х2
: 0 m2 : — ,; S2 : ,;
предельные значения парциальных внутр. энергии, энтальпии, объема растворенного
в-ва конечны.
Для бесконечно разб. р-ров
выполняется ряд простых количеств. закономерностей, к-рые получили назв. законов
разб. р-ров. Это закон Рауля (11) для р-рителя и Генри закон для растворенного
в-ва р2 = Кгх2, где р2
- парциальное давление пара растворенного в-ва, x2-его
молярная доля в р-ре, Кг - постоянная Генри; закон
Вант-Гоффа для осмо-тич. давления p = RTc, где p-осмотич. давление,
с-молярная концентрация растворенного в-ва (см. Осмос); закономерности
понижения т-ры замерзания р-ров и повышения т-ры их кипения, если растворенное
в-во нелетуче. Для данного р-рителя при фиксированных Т и р давление
пара, осмотич. давление, т-ры кипения и замерзания р-ра оказываются зависящими
только от концентрации растворенного в-ва, но не от его природы, что объединяет
названные св-ва в общее понятие коллигативных.
Важной характеристикой
бесконечно разб. р-ров являются функции сольватации-изменения термодинамич.
ф-ций (энергии, энтальпии, стандартной парциальной энтропии, стандартного хим.
потенциала и др.) при переносе растворенного в-ва из идеальной газовой фазы
в бесконечно разб. р-р. Ф-ции сольватации несут количеств. информацию об интенсивности
взаимод. молекул растворенного в-ва с р-ри-телем.
Термодинамические характеристики
неидеальных Р.н.
Неидеальные Р.н. часто
характеризуют т. наз. избыточными термодинамическими функциями МE,
к-рые представляют собой разность между ф-циями смешения реального и идеального
р-ров одинаковой концентрации (рис., б). Обычно эти ф-ции относят к процессу
смешения при р,Т= const, реже-при V, Т= const. С учетом выражений
(10) получаем:
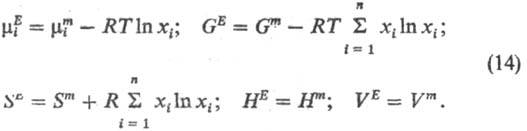
Для представления концентрац.
зависимости хим. потенциала i-го компонента реального р-ра вводят величину
ai, наз. термодинамич. активностью:
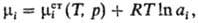
где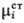 -стандартный
хим. потенциал. В стандартном состоянии ai= 1. Если в качестве
стандартного состояния для всех компонентов принимают соответствующие чистые
жидкости (симметричный способ нормировки), то
-стандартный
хим. потенциал. В стандартном состоянии ai= 1. Если в качестве
стандартного состояния для всех компонентов принимают соответствующие чистые
жидкости (симметричный способ нормировки), то 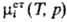 =
=
=
=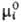 (Т,р).
При нормировке, наз. несимметричной, стандартное состояние для р-рителя,
как и в предыдущем случае,-чистая жидкость, но для растворенного в-ва (компонент
2)
(Т,р).
При нормировке, наз. несимметричной, стандартное состояние для р-рителя,
как и в предыдущем случае,-чистая жидкость, но для растворенного в-ва (компонент
2)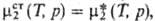 причем в
шкале молярных долей величина
причем в
шкале молярных долей величина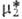 определена
так же, как в ф-ле (13). Стандартное состояние в шкалах молярности и моляльности
отвечает соответствующим единичным концентрациям.
определена
так же, как в ф-ле (13). Стандартное состояние в шкалах молярности и моляльности
отвечает соответствующим единичным концентрациям.
Коэффициенты активности
gi = ai/xi = 1 при
симметричном способе нормировки для чистых жидкостей (xi = 1).
При несимметричном способе нормировки коэффициенты активности и р-рителя и растворенного
в-ва равны единице в бесконечно разб. р-ре: g1 (х2=
0) = g2(x2 = 0) = 1. В случае симметричной
нормировки
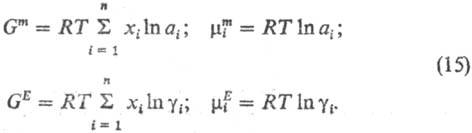
В зависимости от знака
избыточной энергии Гиббса GE говорят о положит. или отрицат.
отклонениях от идеального поведения.
При анализе концентрац.
зависимостей хим. потенциалов (активностей, коэф. активностей) важную роль играет
Гиббса -Дюгема уравнение. В соответствии с этим ур-нием при Т, р =
const
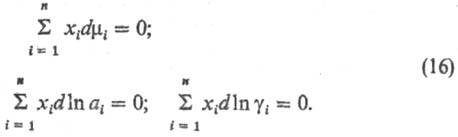
Условия устойчивости р-ра
относительно непрерывных изменений параметров состояния требуют, чтобы при заданных
Тир кривизна пов-сти G(x1, ..., хn-1)
была положительной. Для бинарного р-ра это сводится к условию: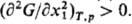 Если
неравенство выполняется во всей области составов, жидкости обладают полной взаимной
р-римостью. Если в к.-л. области составов
Если
неравенство выполняется во всей области составов, жидкости обладают полной взаимной
р-римостью. Если в к.-л. области составов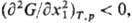 взаимная р-римость ограничена, наблюдается расслаивание р-ра. На границе устойчивости
р-ра относительно непрерывных изменений параметра состояния (на спинодали) В
критич. точке р-римости бинарной системы
взаимная р-римость ограничена, наблюдается расслаивание р-ра. На границе устойчивости
р-ра относительно непрерывных изменений параметра состояния (на спинодали) В
критич. точке р-римости бинарной системы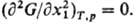 выполняется
записанное равенство, а также равенство
выполняется
записанное равенство, а также равенство 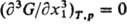 (см. Критическое состояние).
(см. Критическое состояние).
Межмолекулярные взаимодействия
в растворах. Особенности концентрационных и температурных зависимостей термодинамич.
св-в р-ров определяются характером межмолекулярных взаимодействий между одноименными
и разноименными частицами: природой и интенсивностью сил притяжения (дисперсионных,
индукц., ориентац., донорно-ак-цепторных), размерами и формой молекул, т.е.
видом потенциала отталкивания. Учет размеров молекул оказывается чрезвычайно
существенным для объяснения св-в растворов полимеров; далее речь пойдет
гл. обр. о р-рах низко-мол. в-в.
С учетом характера сил
притяжения можно выделить след, типы бинарных р-ров: 1) р-ры, образованные неполярными
компонентами (смеси сжиженных благородных газов, мн. двухатомных газов, смеси
углеводородов, перфторугле-водородов); 2) р-ры, содержащие полярный и неполярный
компоненты; 3) р-ры, образованные двумя полярными компонентами. Кроме того,
каждый из этих типов обнаруживает большое разнообразие в мол. св-вах и термодинамич.
поведении. К системам второго и третьего типа относят, в частности, ассоциир.
р-ры, в к-рых специфич. взаимод. приводят к образованию мол. комплексов из одинаковых
молекул (ассоциаты) или разных (сольваты). Наиб. распространенным случаем специфич.
взаимод. в Р. н. является водородная связь.
Ассоциаты из одинаковых
молекул Аi (i = 2, 3 ...) образуются в том случае,
если молекула А содержит и электро-нодонорные (протоноакцепторные) и электроноакцепторные
(протонодонорные) группы (спирты, карбоновые к-ты и др.). Если молекулы В содержат
такие группы (хотя бы одного типа), молекулы А образуют с ними смешанные ассоциаты,
если не содержат, то ассоциаты не образуются. Если молекула А является только
донором электронов, а молекула В-только акцептором, в чистых жидкостях ассоциаты
отсутствуют, но в р-ре образуются смешанные ассоциаты. Пример такой системы-р-р
ацетон-хлороформ. Среди ассоциир. систем особыми св-вами обладают вода и водные
р-ры, что в большой степени определяется специфич. структурой воды, наличием
пространств. сетки водородных связей, к-рая изменяется под влиянием молекул
растворенных в-в.
Для р-ров, образованных
двумя неполярными (или двумя полярными) компонентами, могут наблюдаться и положит.
и отрицат. отклонения от идеальности, причем положительные встречаются чаще;
для нек-рых систем они весьма велики
(алкан - перфторалкан, вода-кетон и др.). Для смесей полярного и неполярного
компонентов характерны положит. отклонения от идеальности, нередко наблюдается
очень малая взаимная р-римость жидкостей (напр., в системах вода-алкан); именно
к этому типу относят системы с наиб. отклонениями от идеальности. В ассоциир.
р-рах знак и величина избыточных термодинамич. ф-ций определяются в первую очередь
изменением числа специфич. связей в р-ре по сравнению с чистыми жидкостями.
Определяющая роль хим. взаимод. подчеркивалась Д. И. Менделеевым в его учении
о р-рах.
Молекулярная теория
растворов. Структура р-ров отличается от структуры чистой жидкости тем,
что наряду с локальными неоднородностями плотности и распределения молекул по
ориентациям (последнее-в случае нецентральных межмол. сил) в р-рах имеются также
концентрац. неоднородности. Локальные корреляции в расположении молекул и их
ориентации в р-ре м. б. описаны с помощью мол. ф-ций распределения. В бинарной
системе (компоненты 1 и 2) для характеристики распределения частиц в
окрестности нек-рой данной частицы на расстоянии r от нее служат радиальные
функции распределения g11(r), g22(r),
g12(r) = g21(r). Через
эти ф-ции можно выразить локальный состав окружения частицы данного сорта. Для
систем, между молекулами к-рых действуют нецентральные силы, вводятся корреляционные
функции, зависящие от угловых переменных, т.е. от взаимной ориентации молекул.
Статистическая термодинамика
дает принципиальную возможность вычислить структурные и термодинамич. св-ва
системы исходя из ее мол. характеристик и потенциалов межмол. взаимодействия.
Для р-ров, как и для чистых жидкостей, развиваются: 1) аналит. теории, в к-рых
связь между корреляц. ф-циями и потенциалом взаимод. получают в виде интегральных
ур-ний; 2) методы численного моделирования - Монте-Карло и мол. динамики (см.
Молекулярная динамика); 3) возмущений теория; 4)
приближенные модельные, в частности решеточные, теории (см. Жидкость).
Теория разб. р-ров Майера-Макмиллана
на строгой мол.-статистич. основе рассматривает разложение термодинамич. ф-ций
по степеням концентрации или активности растворенного в-ва, дает мол. интерпретацию
коэф. разложений, устанавливает аналогию в разложениях для разб. р-ров и для
газов (по степеням давления). Помимо подхода, опирающегося на информацию о потенциалах
взаимод. и общие идеи мол.-статистич. рассмотрения, для установления связи между
термодинамич. и мол. св-вами разб. р-ров развиваются методы, рассматривающие
р-ритель как непрерывную среду и применяющие к р-ру представления теории диэлектриков.
Мол.-статистич. теории,
ставящие своей задачей вывести структурные и термодинамич. св-ва р-ра из потенциала
взаимод. (т. наз. строгие теории), в последние десятилетия достигли больших
успехов. Для совр. работ в этой области характерен переход от изучения смесей
простых жидкостей (систем с центральными взаимод.) к изучению смесей молекулярных
флюидов, т.е. систем, образованных двух-и многоатомными молекулами, где взаимод.
обычно носят нецентральный характер из-за асимметрии мол. формы (потенциала
отталкивания), наличия электрич. моментов молекул (дипольного, квадрупольного
и др.). Большую роль в исследовании мол. флюидов играют теория возмущений и
численное моделирование. При этом теория возмущений в большой степени опирается
на результаты, полученные для простых систем, в частности для смесей частиц,
моделируемых твердыми сферами разного размера, св-ва к-рых хорошо изучены с
помощью интегральных ур-ний и численными методами. Развиваются варианты теории
возмущений с применением принципа соответств. состояний (теория конформных р-ров).
Исследования, базирующиеся
на интегральных ур-ниях, теории возмущений, численном моделировании, относятся
в осн. к бинарным р-рам. Для бинарных р-ров с центральными взаимод. при небольших
различиях в параметрах потенциалов
взаимод. 1-1 и 2-2 теория возмущений дает зависимости избыточных термодинамич.
ф-ций от состава, близкие к регулярным, т. е. симметричные по компонентам 1
и 2. В частности,
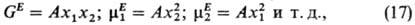
где А -коэф., выражаемый
через св-ва стандартной жидкости и параметры потенциалов взаимод. 1-1, 2-2,
1-2.
Приближенные теории р-ров
исходят из упрощающих допущений относительно структуры р-ра. Эти теории обычно
оперируют параметрами, для определения к-рых требуется эксперим. термодинамич.
информация об исследуемой системе. Теория регулярных растворов Скетчарда-Гильдебрандта
предложена для р-ров, образованных неполярными жидкостями, молярные объемы к-рых
одного порядка (различия не более чем в 2-3 раза). Делается допущение об идеальном
значении энтропии смешения при постоянном объеме. Избыточная внутр. энергия
определяется выражением:
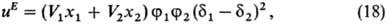
где Vi-молярный
объем i-го компонента (i = 1,2), xi-его молярная
доля,-объемная доля, di =
(Eiисп/Vi)1/2 параметр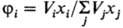 растворимости
(осн.параметр теории), Eiисп-энергия испарения
жидкости.
растворимости
(осн.параметр теории), Eiисп-энергия испарения
жидкости.
Решеточные теории основаны
на допущении о квази-кристаллич. структуре р-ра, хотя, вообще говоря, при этом
преувеличивается аналогия между жидкостью и твердым телом, поскольку жидкости
приписывается дальний порядок. Теория строго регулярных р-ров, развитая в работах
Э. А. Гуггенгейма и др., относится к системам, образованным частицами примерно
одинакового размера, взаимодействующим посредством центр. сил. Осн. параметр
тео-рии-энергия взаимообмена
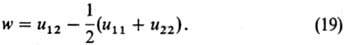
Величина w характеризует
различие между энергией взаимод. пары ближайших соседей 1-2 и средним
арифметическим энергии однотипных взаимод.:
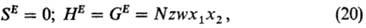
где z-координац.
число решетки, N- общее число частиц в системе. Модель находит применение
в исследованиях твердых сплавов, расплавов.
Модели атермич. р-ров учитывают
влияние размеров молекул на термодинамич. ф-ции, но предполагают, что различия
в энергетике однотипных и смешанных взаимод. отсутствуют (НЕ =
0; GE = — TSE). В приближении Флори
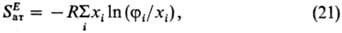
где fi-объемная
доля компонента i, SE- молярная избыточная энтропия;
SE > 0. Приближение Флори широко используется в теории
р-ров полимеров. Развиты подходы, принимающие во внимание не только "объем"
молекул, но и их форму.
Наиб. общие решеточные
модели учитывают факторы размера и формы молекул, а также энергетич. вклады.
При моделировании р-ров с ориентац. эффектами и ассоциацией молекул предполагается,
что энергия взаимод. зависит от взаимной ориентации молекул, способа их контактирования.
Для контактов разл. типа вводятся разл. энергии взаимообмена (Дж. Баркер, 1953).
Решеточные модели, допускающие наличие вакансий в квазирешетке (т. наз. дырочные
модели), позволяют рассматривать объемные эффекты в р-рах, получать ур-ние состояния,
описывающее не только жидкую, но и паровую фазу. В применении к р-рам развиваются
и разл. варианты ячеечных теорий, в к-рых рассматриваются зависимости своб.
объема от состава. Для описания ассоциир. р-ров широко применяют теории ассоциативных
равновесий, представляющие р-р как смесь мономерных частиц,
ассоциатов и сольватов, равновесие между к-рыми определяется действующих
масс законом. Смесь таких мол. образований описывается в том или ином приближении,
относящемся к системе без специфич. взаимод. (самое грубое приближение-идеальная
ассоциир. смесь, затем идет приближение атермич. смеси ассоциатов и т.д.).
В практич. расчетах термодинамич.
св-в Р. н., в частности при моделировании фазовых равновесий жидкость-пар, широкое
распространение получили т. наз. ур-ния локального состава-ур-ние Вильсона,
модели НРТЛ, ЮНИКВАК и др. Для ориентировочного предсказания св-в Р. н. полезны
групповые модели, основанные на допущении об аддитивности вкладов разл. групп
в избыточные термодинамич. ф-ции (модели ЮНИФАК, АСОГ, квазихимические групповые
и др.). В настоящее время развиты модификации моделей, дающие возможность получить
ур-ние состояния жидкой и паровой фаз, что особенно важно при расчетах фазовых
равновесий в широком диапазоне условий.
80-е гг. 20 в. отмечены
успехами в области мол. теории неравновесных св-в р-ров, но трудности здесь
еще более серьезные, чем при изучении равновесных св-в.
Лит.: Пригожин И.,
Дефэй Р., Химическая термодинамика, пер. с англ., Новосиб., 1966; Фиалков Ю.
А., Житомирский А.Н., Тарасенко Ю.А., Физическая химия неполных растворов, Л.,
1973; Шахпаронов М. И., Введение в современную теорию растворов, М., 1976; Герасимов
Я. И., Гейдерих В. А., Термодинамика растворов, М., 1980; Термодинамика разбавленных
растворов неэлектролитов, Л., 1982; Белоусов В. П., Панов М. Ю., Термодинамика
водных растворов неэлектролитов, Л., 1983; Смирнова Н.А., Молекулярные теории
растворов, Л., 1987; Пригожий И. Р., Молекулярная теория растворов, пер. с англ.,
М., 1990. Н. А. Смирнова.