


ТИТРИМЕТРИЯ (от
франц. titre- качество, характеристика и греч. metreo-измеряю), совокупность
методов количеств. анализа, основанных на измерении кол-ва реагента, необходимого
для взаимод. с определяемым компонентом в р-ре или газовой фазе в соответствии
со стехиометрией хим. р-ций между ними. При проведении эксперимента можно контролировать
либо объем, либо массу добавляемого титранта-р-ра или газовой смеси с точно
известной концентрацией Ст реагента. Наиб. распространение
получила Т. для экспрессного определения высоких и средних концентраций в-в
в р-рах, в т. ч. неводных. Точно известный объем V анализируемого р-ра
с помощью пипетки помещают в конич. колбу и к нему прибавляют небольшими порциями
титрант из бюретки (калибров. стеклянная трубка с клапанным устройством, напр.
краном, на оттянутом конце), тщательно перемешивая р-р в колбе. Эту операцию
наз. титрованием. Массовые титриметрич. анализы обычно проводят с помощью титраторов.
Процесс титрования сопровождается
изменением равновесных концентраций реагента, определяемого в-ва и продуктов
р-ции. Это удобно изобразить графически в виде т. наз. кривой титрования в координатах
концентрация опре-деляемого.в-ва (или пропорциональная ей величина)-объем (масса)
титранта.
Измерив объем Vт
титранта в конце титрования, рассчитывают концентрацию С анализируемого
р-ра по формуле: CV= CтVт (концентрации
выражены в моль/л). Теоретически необходимо добавить такой объем титранта, к-рый
содержит кол-во реагента, эквивалентное кол-ву определяемого компонента в соответствии
со стехиометрией р-ции между ними при условии, что эта р-ция практически необратима.
Этот объем титранта соответствует точке эквивалентности (т. э.), или моменту
стехиометричности. Практически определяют, однако, не т.э., а конечную точку
титрования (к.т.т.), к-рая должна максимально совпадать с т.э. для получения
миним. погрешности титрования. Фиксировать к.т.т. можно по изменению окраски
добавленного индикатора (выбор к-рого осуществляют по теоретически рассчитанной
кривой титрования) или по достаточно резкому изменению к.-л. физ. характеристики
р-ра, зависящей от концентрации определяемого в-ва,-тока, окис-лит.-восстановит.
потенциала, оптич. плотности, электрич. проводимости и кол-ва электричества.
Соотв. различают амперометрическое титрование, потенциометрич., фотометрич.,
кондуктометрич. и кулонометрич. титрование (см. Потенциометрия, Фотометрический
анализ, Кон-дуктометрия и Кулонометрия). В этих титриметрич.
методах кривая титрования представляет собой зависимость измеряемой физ. величины
от объема (или массы) титранта.
Часто строят логарифмич.
кривую титрования, откладывая по оси ординат значения lgC (или величины,
пропорциональные им), а по оси абсцисс-степень оттитрованности f (безразмерная
величина или в %), к-рая равна отношению кол-ва nт (или объема
Vт) добавленного титранта к кол-ву nт.э.
(или объему Vт.э.) титранта, необходимому для достижения т.э.:
f= Vт/Vт.э. = nт/nт.э..
Тогда до т.э. f< 1 (<100%), в т.э. f = 1 (100%), а после
т.э. f> 1 (>100%). Соотв. логарифмич. кривая титрования состоит
из трех участков: пологая ветвь, крутой подъем (скачок титрования) и вторая
пологая ветвь (см. рис.). Величина скачка титрования зависит от константы равновесия
Кр р-ции между определяемым в-вом и титрантом (р-ция
титрования). С уменьшением величины lgKp скачок уменьшается.
Если при выполнении анализа допустима погрешность порядка b0,1%, за начало
скачка принимают точку f = 0,999, а за его конец-точку f = 1,001.
В этом случае только при lgKp > 6 погрешность из-за несовпадения
к. т. т. и т. э. не превысит допустимую.
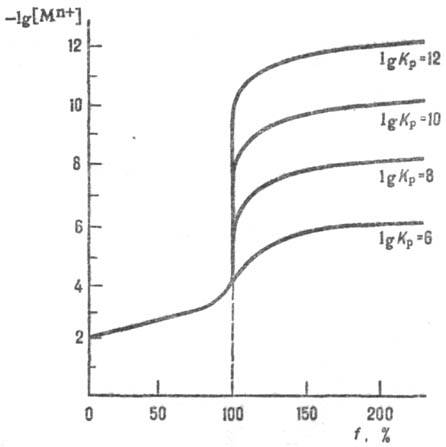
Логарифмич. кривые титрования
р-ра ионов металлов Мn+ р-ром этилен-диаминтетрауксусной
к-ты Н4Y; Кp - константа равновесия р-ции
титрования Mn++Y4-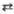 MYn-4;
Kp=[MYn-4]/[Mn+][Y4-].
MYn-4;
Kp=[MYn-4]/[Mn+][Y4-].
В Т. применяют разл. хим.
р-ции: кислотно-основные (см. Кислотно-основное титрование), окислит.-восстановит.,
комплексообразования, а также т. наз. р-ции осаждения, приводящие к образованию
осадка. Р-ция титрования должна протекать не только стехиометрически, но также
быстро и количественно (правильные результаты можно получить лишь в том случае,
если при прибавлении стехио-метрич. кол-ва титранта полнота протекания р-ции
не менее 99,9%). Кроме того, в каждом конкретном случае должен быть подходящий
способ фиксирования к.т. т.
Окислит.-восстановит. титрование
основано на р-циях типа: aOx1 + bRed2 +
...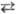 bОx2
+ aRed1 + ..., где Ox1 и Ох2 -
окисленные формы соотв. титранта и определяемого в-ва, Red1 и Red2-их
восстановл. формы, a, b, a, b-стехио-метрич. коэффициенты.
Окислит.-восстановит. потенциалы, характеризующие титрант и определяемое в-во,
соотв. имеют вид:
bОx2
+ aRed1 + ..., где Ox1 и Ох2 -
окисленные формы соотв. титранта и определяемого в-ва, Red1 и Red2-их
восстановл. формы, a, b, a, b-стехио-метрич. коэффициенты.
Окислит.-восстановит. потенциалы, характеризующие титрант и определяемое в-во,
соотв. имеют вид:
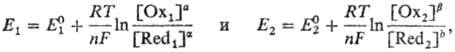
где 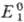 и
и 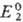 -реальные
потенциалы пар соотв. Ox1/Red1 и Ox2/Red2
в данной системе, n-число электронов, участвующих в р-ции, F- число
Фарадея, R-универс. газовая постоянная. Титрант действует как окислитель,
если
-реальные
потенциалы пар соотв. Ox1/Red1 и Ox2/Red2
в данной системе, n-число электронов, участвующих в р-ции, F- число
Фарадея, R-универс. газовая постоянная. Титрант действует как окислитель,
если 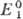 >
>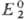 ,
и как восстановитель, если .
,
и как восстановитель, если .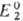 >
>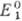 .
В ходе титрования величина |Е1—Е2| уменьшается,
а в т. э. Е1 = Е2. Титрование
эффективно, если при этом отношение [Red2]/[Ox2] (в случае
титрования окислителем) или обратное
соотношение (в случае титрования восстановителем) мало. Для этого подбирают
титрант так, чтобы
.
В ходе титрования величина |Е1—Е2| уменьшается,
а в т. э. Е1 = Е2. Титрование
эффективно, если при этом отношение [Red2]/[Ox2] (в случае
титрования окислителем) или обратное
соотношение (в случае титрования восстановителем) мало. Для этого подбирают
титрант так, чтобы было достаточно велико. В соответствии с тем, какое в-во служит титрантом, различают
перманганатометрию, иодо-метрию, дихроматометрию, броматометрию и т.
д. Конечную точку устанавливают, как правило, потенциометри-чески или с помощью
окислит.-восстановит. индикаторов, а также по появлению или исчезновению окраски
титранта или титруемого в-ва.
было достаточно велико. В соответствии с тем, какое в-во служит титрантом, различают
перманганатометрию, иодо-метрию, дихроматометрию, броматометрию и т.
д. Конечную точку устанавливают, как правило, потенциометри-чески или с помощью
окислит.-восстановит. индикаторов, а также по появлению или исчезновению окраски
титранта или титруемого в-ва.
Метод, основанный на образовании
устойчивых комплексных соед. катионов металлов с разл: лигандами, наз. комплексометрией.
Конечную точку устанавливают с помощью металлохромных индикаторов или в-в, взаимодействующих
с избытком реагента (лиганда). К комплексо-метрии относятся, в частности, комплексонометрия,
методы, основанные на образовании иодидных комплексов Hg (см. Меркуриметрия),
фторидных комплексов Ag и Zr (фториметрия).
Примерами осадит. титрования
могут служить методы определения галогенидов с применением солей серебра (см.
Аргентометрия), а также сульфатов с помощью р-римых солей Ва.
В последнем случае осаждение BaSO4 ведут в смеси воды и метанола
(1:1); индикатором служит ализариновый красный С, меняющий окраску при адсорбции
на пов-сти осадка.
Во всех титриметрич. методах
р-р титранта лучше всего готовить из т. наз. первичного стандарта-химически
чистого, устойчивого в твердом виде и в р-ре в-ва, напр. Na2CO3
или К2Сr2О7. В этом случае р-р с известной
концентрацией (т. наз. стандартный р-р) получают, растворяя точную навеску этого
в-ва в мерной колбе (см. также Фиксаналы). Однако многие широко
используемые титран-ты не отвечают этим требованиям. Напр., Na2S2O3
в р-ре разлагается вследствие взаимод. с растворенными О2 и СО2,
a NaOH (или КОН) не является химически чистым, т.к. содержит переменные кол-ва
карбонатов. В таких случаях готовят р-р титранта приблизительно требуемой концентрации,
а затем точно определяют ее (стандартизируют р-р) с помощью подходящего первичного
стандарта.
По технике выполнения различают
прямое, обратное и заместительное (косвенное) титрование. При прямом титровании
р-р титранта добавляют непосредственно к р-ру определяемого компонента. Если
скорость р-ции титрования мала или нет подходящего индикатора, то прибегают
к обратному титрованию. Для этого к анализируемому р-ру прибавляют избыток титранта
и по окончании р-ции (если она идет очень медленно, то р-р иногда нагревают)
избыток титранта оттитровывают р-ром др. подходящего реагента. Косвенным титрованием
пользуются, если определяемое в-во с данным титрантом не реагирует или реагирует
нестехиометрически. В этом случае к анализируемому р-ру прибавляют вспомогат.
реагент, с к-рым определяемое в-во образует стехиометрич. кол-во нового соед.
(заместителя), к-рое затем определяют прямым титрованием. Напр., Na2S2O3
с сильными окислителями (К2Сr2О7, КIO3,
КВrO3) реагирует нестехиометрически, поэтому для определения К2Сr2О7
к анализируемому р-ру добавляют избыток KI и кол-во выделившегося заместителя
I2 определяют прямым титрованием р-ром Na2S2O3.
Т. возникла в сер. 18 в.
Многие ученые внесли вклад в ее развитие. Так, У. Льюис (1767) дал определение
понятия "точки насыщения", т. е. точки эквивалентности. Благодаря
работам Ж. Гей-Люссака Т. превратилась из метода пром. анализа в самостоят.
раздел науки. Э. Мор разработал много методик по титриметрич. анализу, написал
"Учебник по химико-аналитическому методу титрования" (1856); В.
Оствальд и А. Ганч развили теорию индикаторов (1894); Д. Форлендер впервые провел
титрование в неводных средах (1904).
Т. отличается малой трудоемкостью,
простотой аппаратурного оформления; довольно высокой точностью и широко применяется
при научных исследованиях и при контроле технол. процессов.
Лит.: Скуг Д., Уэст
Д., Основы аналитической химии, пер. с англ., т. 1, М., 1979; Мейтис Я., Введение
в курс химического равновесия и кинетики, пер. с англ., М., 1984. Г. В. Прохорова.