


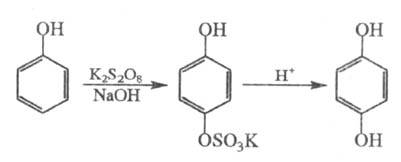
ЭЛЬБСА РЕАКЦИИ, 1) получение двухатомных фенолов действием персульфатов на одноатомные фенолы в щелочной среде (окисление по Эльбсу, персульфатное окисление), напр.:
Электронодонорные заместители ускоряют,
а электроноакцепторные замедляют р-цию. Вторая группа НО обычно вступает
в пара-положение (если оно занято, то образуются opmo-дифенолы,
но выход резко снижается). Гладко в Э. р. вступают крезолы, n-галоген-,
о-
и n-нитрофенолы, м-гидроксибензальдегид и м-гидроксибензойная
к-та. Окисление двухатомных фенолов часто сопровождается разрушением ароматич.
ядра. Если одну из групп НО предварительно проалкилировать, то р-ция протекает
обычным образом.
 -Нафтол и его производные дают с хорошими выходами 1,4-дигидроксипроизводные,
-Нафтол и его производные дают с хорошими выходами 1,4-дигидроксипроизводные, -нафтол
и 2-гидрокси-З-нафтойная к-та образуют 1,2-дигидроксипроизводные, но с
низкими выходами.
-нафтол
и 2-гидрокси-З-нафтойная к-та образуют 1,2-дигидроксипроизводные, но с
низкими выходами.
Р-цию обычно осуществляют добавлением
на холоде насыщ. водного р-ра K2S2O8 или
(NH4)2S2O8 к щелочному р-ру фенола с послед.
подкислением. Использование смеси K2S2O8
и
ацетата Pd позволяет вести прямое ацетоксилирование одноатомных фенолов.
Выходы, как правило, не превышают 50%.
Осн. побочные продукты - изомерные дигидроксифенолы
(характерны для р-ции замещенных фенолов), хиноны и продукты их дальнейшей
деструкции.
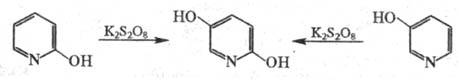
В р-цию, подобную Э. р., также вступают
гидроксипроизводные флавона, кумарина, нек-рых азотсодержащих гетероциклов,
напр.:
Механизм р-ции окончательно не выяснен.
Предполагают, что она может осуществляться по свободнорадикальному механизму
(следы Fe2+ в персульфате катализируют образование анион-радикала
SO-4, к-рый атакует пара-положение фенолят-иона)
или ионному (атака феноксид-аниона ионом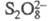 и послед. перегруппировка образовавшегося аддукта).
и послед. перегруппировка образовавшегося аддукта).
Э. р. используют в препаративной практике;
открыта К. Эльбсом в 1893.
Лит.: Sethna S., "Chem. Revs.", 1951, № 49, № 1, p. 91-101; Sheldon R., Koschi J., Metall-catalized oxidation of organic compounds, N. Y., 1981; Capdevielle P., Maumy M., "Tetrahedron letters", 1982, v. 23, № 15, p. 1573-76, 1577-80.
2) Термич. циклодегвдратация диарилкетонов, содержащих в орто-положении группы СН3 или СН2, с образованием конденсир. аромстич. углеводородов, напр.:
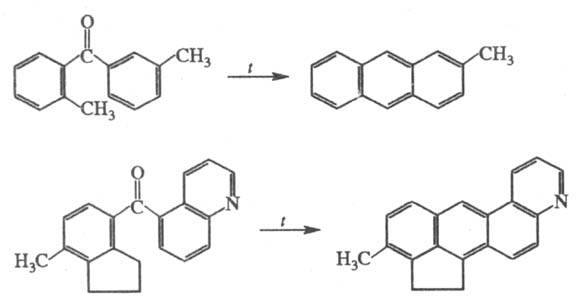
Р-цию обычно осуществляют при 350-500 °С
в течение неск. часов или суток до прекращения выделения Н2О.
Выходы обычно не превышают 50%.
Осн. побочные процессы: миграция карбонильной
группы и др. внутримол. перегруппировки; отщепление заместителей (Аlk,
АlkО, Hal) из ароматич. ядра; гидро- и дегидрогенизация.
Механизм р-ции не выяснен. Полагают, что
первоначально кетон переходит в енольную форму с образованием сопряженной
системы связей; затем следует внутримол. присоединение др. ароматич. кольца
с образованием производного дигидроантранола, к-рый подвергается дегидратации.
Существуют и др. представления о механизме этой р-ции.
Р-цию циклодегидратации используют в препаративной
практике как наиб. экономичный и быстрый способ синтеза нек-рых производных
дибензо[а,h]антрацена, холантрена и динафтохризенов.
Р-ция открыта К. Эльбсом в 1884.
Лит.: Физер Л., в сб.: Органические реакции, пер. с англ., сб. 1, М., 1948, с. 163; Фьюзон Р., Реакции органических соединений, пер. с англ., М., 1966, с. 450-51.
Г. И. Дрозд.